Biochemical Genetics Department
ΝΟΣΟΣ POMPE, Η άμεση διάγνωση παραμένει σημαντική

Η 15η Απριλίου έχει καθιερωθεί ως η Παγκόσμια Ημέρα Ενημέρωσης και Ευαισθητοποίησης για τη Νόσο Pompe. Η νόσος Pompe οφείλει το όνομά της στον Ολλανδό παθολόγο Joannes Cassianus Pompe, ο οποίος περιέγραψε πρώτος σε δημοσίευσή του το 1932 την περίπτωση ενός κοριτσιού, ηλικίας 7 μηνών, που πέθανε αιφνίδια από υπερτροφία της καρδιάς και στην οποία παρατηρήθηκε μαζική συσσώρευση γλυκογόνου εντός κενοτοπίων, που αργότερα χαρακτηρίστηκαν ως λυσοσώματα.
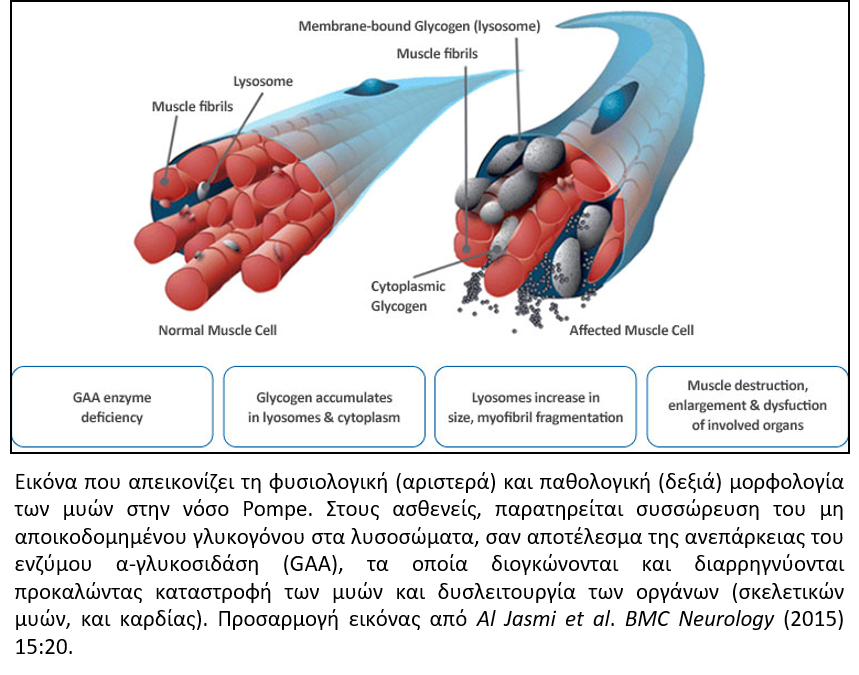
Η νόσος Pompe ή γλυκογονίαση τύπου ΙΙ (Glycogen storage disease type II) είναι μια σπάνια κληρονομική μεταβολική μυοπάθεια που ανήκει στην κατηγορία των μεταβολικών ασθενειών των λυσοσωμάτων. Εμφανίζεται με συχνότητα περίπου 1/40.000 γεννήσεις, σύμφωνα με τη διεθνή βιβλιογραφία και κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο. Προκαλείται από αλλοιώσεις στο γονίδιο που ευθύνεται για την παραγωγή της α-γλυκοσιδάσης (όξινης μαλτάσης), του ενζύμου που είναι υπεύθυνο για την διάσπαση του γλυκογόνου σε γλυκόζη στα λυσοσώματα. Ανεπάρκεια του συγκεκριμένου ενζύμου προκαλεί συσσώρευση του γλυκογόνου εντός των λυσοσωμάτων σε διάφορους ιστούς, αλλά κυρίως στους σκελετικούς και καρδιακούς μύες.
Η νόσος Pompe χαρακτηρίζεται από ένα ευρύ φάσμα κλινικών εκδηλώσεων και ταξινομείται ανάλογα με την ηλικία έναρξης, τη σοβαρότητα και το ρυθμό εξέλιξης των συμπτωμάτων. Διακρίνονται δύο εκδηλώσεις της νόσου, η κλασική πρώιμη μορφή, όπου τα συμπτώματα εμφανίζονται εντός των πρώτων μηνών της ζωής και η όψιμη εκδήλωση της νόσου, στην οποία τα συμπτώματα εμφανίζονται σε μετέπειτα στάδιο (παιδική έως ενήλικη ζωή) και εξελίσσονται με βραδύτερο ρυθμό. Η κλινική ετερογένεια της νόσου, ως επί το πλείστον, οφείλεται στο διαφορετικό ποσοστό ενεργότητας του ενζύμου α-γλυκοσιδάση που προκύπτει ανάλογα με τον τύπο της γενετικής αλλοίωσης στο αντίστοιχο γονίδιο (GAA). Η κλασική μορφή της νόσου χαρακτηρίζεται από σχεδόν πλήρη έλλειψη ενεργότητας του ενζύμου και εκδηλώνεται εντός των πρώτων μηνών της ζωής με καρδιομεγαλία, υποτονία, ταχέως εξελισσόμενη μυϊκή αδυναμία, ηπατομεγαλία, μακρογλωσσία, δυσκολία στη σίτιση και αναπνευστική ανεπάρκεια. Η πορεία της νόσου σε αυτές τις περιπτώσεις είναι ταχέως προοδευτική και χωρίς θεραπευτική παρέμβαση ο θάνατος επέρχεται εντός του πρώτου/δεύτερου έτους της ζωής, λόγω καρδιοαναπνευστικής ανεπάρκειας. Οι ασθενείς με την όψιμη μορφή παρουσιάζουν μερική ενεργότητα του ενζύμου και βραδύτερη εξέλιξη των συμπτωμάτων τα οποία δεν περιλαμβάνουν καρδιομυοπάθεια.
Η θεραπεία ενζυμικής υποκατάστασης για τη νόσο Pompe είναι διαθέσιμη από το 2006, όπου και εξασφάλισε ευρωπαϊκή άδεια κυκλοφορίας, και άνοιξε νέους ορίζοντες στην αντιμετώπιση της ασθένειας, ιδιαίτερα της κλασικής πρώιμης μορφής της νόσου. Τα αποτελέσματα της συγκεκριμένης θεραπείας υπήρξαν εντυπωσιακά αναφορικά με τη βελτίωση της μυοκαρδιοπάθειας και της καρδιομεγαλίας, μειώνοντας τον κίνδυνο θανάτου κατά 99%. Το μέγιστο θεραπευτικό όφελος για τον ασθενή επιτυγχάνεται με την εφαρμογή της θεραπείας ενζυμικής υποκατάστασης το νωρίτερο δυνατό. Λόγω αυτού προκύπτει η ανάγκη της έγκαιρης διάγνωσης, κάτι που οδήγησε στην εφαρμογή ανιχνευτικών νεογνικών προγραμμάτων για τη νόσο Pompe σε αρκετές χώρες, μέσω της μέτρησης της ενεργότητας της α-γλυκοσιδάσης. Αυτό είχε ως συνέπεια να εντοπίζονται εκτός από τους ασθενείς με πλήρη ανεπάρκεια, άτομα με μερική ενεργότητα του ενζύμου σε ασυμπτωματικό στάδιο, για τα οποία η πορεία της νόσου είναι αβέβαιη και το θεραπευτικό όφελος της ενζυμικής υποκατάστασης δεν είναι ακόμα γνωστό.
Η πρόκληση στη διαχείριση ασυμπτωματικών ατόμων με χαμηλή ενζυμική ενεργότητα αναδεικνύεται σε μια πρόσφατη δημοσίευση του Τμήματος Βιοχημικής Γενετικής στο επιστημονικό περιοδικό Molecular Genetics and Metabolism Reports με τίτλο “GAA variants associated with reduced enzymatic activity but lack of Pompe-related symptoms, incidentally identified by exome sequencing” (DOI: 10.1016/j.ymgmr.2023.100997). Στη συγκεκριμένη μελέτη περιγράφονται ασθενείς στους οποίους ανιχνεύτηκαν γενετικές παραλλαγές στο γονίδιο GAA ως τυχαίο εύρημα της διερεύνησης τους μέσω της τεχνικής αλληλούχισης επόμενης γενιάς (Next Generation Sequencing, NGS), λόγω κλινικών συμπτωμάτων που δε σχετίζονταν με τη νόσο Pompe. Διαπιστώθηκε ότι οι συγκεκριμένες παραλλαγές στο γονίδιο GAA σχετίζονταν με μειωμένη ενζυμική ενεργότητα που θα ήταν ενδεχομένως συμβατή με την όψιμη μορφή της νόσου. Οι ίδιες γενετικές αλλοιώσεις και παρόμοια βιοχημικά ευρήματα ανιχνεύτηκαν σε ασυμπτωματικά μέλη της ίδιας οικογένειας.
Εκτός από τη συσχέτιση του συγκεκριμένου συνδυασμού γενετικών παραλλαγών με τη μειωμένη ενεργότητα της α-γλυκοσιδάσης, η μελέτη αναδεικνύει τον αντίκτυπο των τυχαίων ευρημάτων που προκύπτουν από την τεχνική αλληλούχισης επόμενης γενιάς τόσο στην κλινική διαχείριση των ασθενών όσο και στην οικογένεια γενικότερα. Παρόλα αυτά, με δεδομένες τις υπάρχουσες και μελλοντικές θεραπευτικές προσεγγίσεις η έγκαιρη διάγνωση αυτών των ατόμων παραμένει σημαντική. Για αυτό το λόγο το γονίδιο GAA συγκαταλέγεται ανάμεσα στα γονίδια που το American College of Medical Genetics and Genomics (ACMG) συνιστά να αναφέρονται όταν σε αυτά ανιχνεύονται παραλλαγές, ως τυχαία ευρήματα κατά την τεχνική αλληλούχισης επόμενης γενιάς, σύμφωνα με τις κατευθυντήριες οδηγίες που εξέδωσε.
Το Τμήμα Βιοχημικής Γενετικής του Ινστιτούτου Νευρολογίας & Γενετικής Κύπρου προσφέρει τη μέτρηση της α-γλυκοσιδάσης τόσο σε δείγμα αίματος όσο και σε βιοψία μυός.
Δρ Άννα Μαλέκκου
Associate Scientist
Τμήμα Βιοχημικής Γενετικής






0002.jpg)