Neurogenetics Department
ΕΡΕΥΝΑ, Παθογόνα παραλλαγή γονιδίου CLN6

Νέα παθογόνα παραλλαγή του γονιδίου CLN6 που σχετίζεται με τη νεανική νευρωνική κεροειδή λιποφουσκίνωση σε Κύπριους ασθενείς
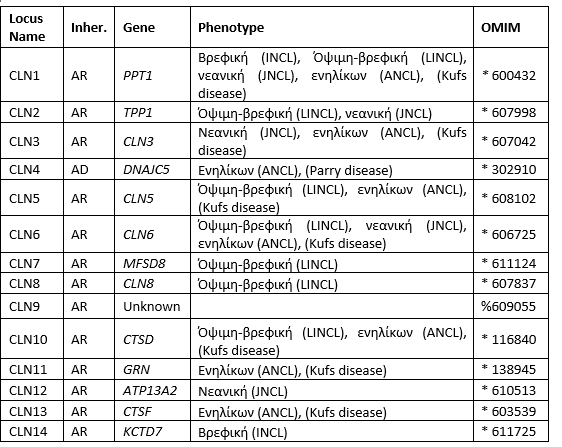
Οι νευρωνικές κεροειδείς λιποφουσκινώσεις (NCLs), είναι μια γενετικά ετερογενής ομάδα αυτοσωμικών υπολειπόμενων διαταραχών λυσοσωμικής αποθήκευσης (υπερβολική συσσώρευση λιποχρωστικών – λιποφουσκίνη). Οι NCLs χαρακτηρίζονται από νευροεκφυλισμό, αταξία, απώλεια όρασης, επιληπτικές κρίσεις και πρόωρο θάνατο. H πρώτη πιθανή περίπτωση της πάθησης αυτής αναφέρθηκαν το 1826 από το Δρ. Christian Stengel, Νορβηγό ιατρό. Σήμερα, παθογόνες παραλλαγές σε περισσότερα από 13 γονίδια έχουν συσχετιστεί με τις NCLs (Πίνακας 1). Η συχνότητα εμφάνισης της νόσου ποικίλλει από τύπο σε τύπο και από χώρα σε χώρα και παρουσιάζεται περίπου σε 1 ασθενή ανά 10,000 πληθυσμού.
Πίνακας 1: Ταξινόμηση των NCLs βάση του υπεύθυνου γονιδίου
Οι μεταλλάξεις στο γονίδιο CLN6 μπορεί να προκαλέσουν διαφορετικές μορφές της νόσου LINCL, JNCL, ΑNCL και Kufs. Το γονίδιο CLN6 κωδικοποιεί την πρωτεΐνη του ενδοπλασματικού δικτύου (ER) η οποία βοηθά τα κύτταρα να απαλλαγούν από τα υλικά που δεν χρειάζονται πλέον.
Τα κλινικά τυπικά συμπτώματα του τύπου αυτού περιλαμβάνουν επιληψία, αταξία, δυσαρθρία, προοδευτική απώλεια νοητικής λειτουργίας και συνήθως μη απώλεια όρασης. Οι ενήλικες με αυτή τη διαταραχή συνήθως δεν επιβιώνουν περισσότερο από 10 χρόνια μετά τη διάγνωση.
Στο τμήμα Νευρογενετικής του Ινστιτούτο Νευρολογίας και Γενετικής Κύπρου διεξάχθηκε έρευνα σε δύο κυπριακές οικογένειες με JNCL, για τον εντοπισμό των παθογόνων μεταλλαγών. Στην παρούσα έρευνα μέλη από τις δύο κυπριακές οικογένειες αξιολογήθηκαν κλινικά και συλλέχθηκαν δείγματα από τα άτομα που συναίνεσαν. Στη συνέχεια έγινε μοριακή διερεύνηση με αλληλούχιση νέας γενιάς (NGS).
Συγκεκριμένα είχαμε τρείς ασθενείς από δύο κυπριακές οικογένειες με νεανική μορφή (JLINCL) (Εικόνα 1). Όλοι οι ασθενείς ήταν αγόρια και τα πρώτα συμπτώματα εμφανίστηκαν στην ηλικία των έξι ετών. Το παιδί της οικογένειας 926 παρουσίασε απώλεια κινητικών ικανοτήτων, αταξία, σπαστικότητα, επιληπτικές κρίσεις και επιληψία. Η μαγνητική τομογραφία εγκεφάλου (MRI) έδειξε υπομυελίνωση. Το παιδί της δεύτερης οικογένειας (915) είχε αταξία, σπαστικότητα, δυσαρθρία, δυστονία και διανοητική αναπηρία. Η MRI έδειξε παρεγκεφαλιδική ατροφία. Κανένα από τα τρία παιδιά δεν εμφάνισε αρχικά σημάδια όρασης ή/και απώλειας ακοής. Οφθαλμολογική αξιολόγηση σε μεταγενέστερο στάδιο έδωσε ενδείξεις μειωμένης οπτικής δραστηριότητας.
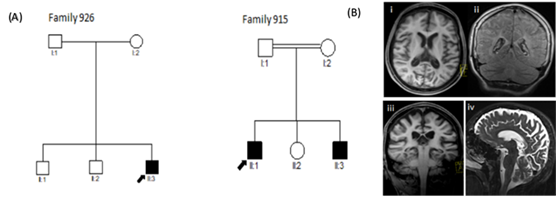
Εικονα 1: Γενεαλογικό δέντρο των οικογενειών (926 & 915). (Β) Αντιπροσωπευτικές αξονικές εικόνες μαγνητικής τομογραφίας (MRI) του εγκεφάλου του ασθενή της οικογένεια 926.
Η μοριακή ανάλυση της οικογένειας 926 έδωσε δύο παραλλαγές (μεταλλάξεις) στο γονίδιο CLN6, την c.407G>A (p.Arg136His) και την c.884A>G (p.Tyr295Cys). Στην οικογένεια 915, και οι δύο ασθενείς ήταν ομόζυγοι για την παραλλαγή c.407G>A (p.Arg136His) (Εικόνα 2Α). Περαιτέρω ανάλυση των παραλλαγών αυτών έδειξε ότι προκαλούν διαφοροποίηση της πρωτεΐνης, είναι παθογόνες και το πιθανότερο η αιτία πρόκλησης της νόσου (Εικόνα 2).
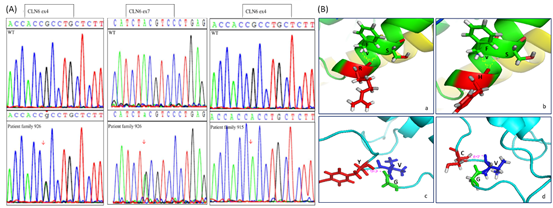
Εικονα 2: (Α) Αλληλουχία των παραλλαγών του γονιδίου CLN6 στις οικογένειες 926 και 915 καθώς και σε υγιή άτομο (WT). (Β) Μοντελοποίηση των παραλλαγών της πρωτεΐνης CLN6 p.Tyr295Cys, και τη p.Arg136His.
Καταληκτικά, στα πλαίσια της μελέτης αυτής περιγράφουμε δύο κυπριακές οικογένειες με νεανικού τύπου JNCL. Και οι δύο οικογένειες είχαν παραλλαγές στο γονίδιο CLN6. Ωστόσο, παρουσίασαν ελαφρώς διαφορετικά κλινικά συμπτώματα. Η ανάλυση πρόβλεψης και δομικής μοντελοποίησης έδειξαν ότι και οι δύο παραλλαγές είναι πιθανότατα παθογόνες. Τα αποτελέσματα αυτά έχουν πρόσφατα δημοσιευτεί στο έγκριτο διεθνές επιστημονικό περιοδικό Frontiers in Genetics (https://doi.org/10.3389/fgene.2021.746101).
Δρ. Πασχάλης Νικολάου
Scientist
Τμήμα Νευρογενετικής
Ινστιτούτο Νευρολογίας και Γενετικής Κύπρου






0002.jpg)







