Molecular Genetics Thalassaemia Department
Haemomics project
Haemomics project – Omic studies of the mechanism of activation of γ-globin for the treatment of haemoglobinopathies
The project aims to the identification of new proteins involved in the regulation of γ-globin gene expression with the intention of identifying suitable pharmacological targets for the treatment of β-haemoglobinopathies (β-thalassaemia and sickle cell disease). Reactivation of γ-globin for the production of fetal haemoglobin (HbF) has long been recognized as a very promising therapeutic approach, since the phenotype of patients with β-thalassaemia or sickle cell disease can be ameliorated. The project is funded by the national Research and Innovation Foundation (RIF) under the RESTART Programs 2016-2020/Excellence Hubs (EXCELLENCE/1216/0389).
β-haemoglobinopathies are the most common genetic disorders worldwide and are inherited in an autosomal recessive manner. β-thalassaemia is caused by reduced or absent expression of β-globin gene whereas sickle cell disease results from a single nucleotide mutation on the same gene. In β-thalassaemia, accumulation of unbound α-globin chains that precipitate in erythroid precursors in the bone marrow and in mature erythrocytes, leads to severe anemia due to ineffective erythropoiesis and peripheral haemolysis. In sickle cell disease, polymerization of sickle haemoglobin at low oxygen levels impairs developing red blood cells leading to loss of their deformability, microvascular damage, anemia due to haemolysis, pain and stroke.
The management of patients with β-haemoglobinopathies is largely supportive rather than curative and includes regular blood transfusions and iron chelation therapy for β-thalassaemia or hydroxyurea treatment among others for sickle cell disease. At the moment, the only curative treatment for these diseases is the allogeneic haematopoietic stem cell transplantation however, this is limited to those having an HLA-matched sibling donor and access to high-technology medicine. β-thalassaemia has been a major health problem in several Mediterranean countries with 12% of Cyprus population being β-thalassaemia carriers. Although the life quality and expectancy of the patients have improved immensely due to improvements in the treatment protocols, patients are far from being morbidity-free.
Pharmacological reactivation of γ-globin for the production of fetal haemoglobin (HbF) is a very promising therapeutic approach for β-thalassaemia and sickle cell disease, as HbF can substitute for the reduced/absent or defective adult haemoglobin (HbA). Chemical agents currently used for this purpose have limited application due to modest therapeutic properties, variable patient response and potential cytotoxic effects. Therefore, identification of novel HbF-inducing agents is of critical importance.
The overall aim of the project is the performance of a CRISPR/Cas9 knockout screen for the discovery of novel γ-globin repressors, which can be targeted pharmacologically for the treatment of β-haemoglobinopathies. Following the identification of a promising candidate, this gene will be validated for the phenotype of interest, that is the upregulation of γ-globin and further subjected to functional analysis. Functional experiments will include transcriptomics and mass spectrometry analysis. These techniques will allow the identification of the effect that this gene’s knockout has at the transcriptome level together with the discovery of interacting protein partners. In the case that the gene of interest is part of a complex affecting transcription or is itself a transcription factor, chromatin immunoprecipation coupled with next-generation sequencing (ChIP-seq) will be performed for the identification of its genome-wide binding sites. These experiments will provide insights into the molecular mechanism by which this gene modulates γ-globin gene expression. The information generated from this project will be useful both because it will increase the available knowledge on the regulation of γ-globin expression but more practically because it can identify novel potential pharmacological targets that can be manipulated to elevate HbF levels in patients.
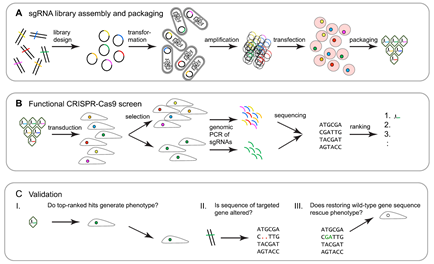
Workflow of CRISPR-Cas9 functional genetic screens. (A) sgRNA libraries are ligated onto plasmid backbones and are then transformed into electrocompetent bacterial cells. The amplified sgRNA library is purified from a bacterial lysate and transfected into virus-producing cells to generate a sgRNA library. (B) The sgRNA library is transduced into target cells, which are subsequently subjected to phenotype selection. Genomic DNA is then harvested, and embedded sgRNAs are amplified by PCR and identified by NGS. Hits are determined and ranked by their relative enrichment or depletion of the respective sgRNAs in the selected versus non-selected control cells. (C) The initial validation of screen hits typically relies on: I. small-scale repeat analysis targeting genes of interest with sgRNAs that had been used in the original screen, plus additional sgRNAs directed toward the same gene; II. genomic sequencing-based verification that the targeted gene was indeed sequence-altered; and III. verification that restoring the wild-type gene sequence rescues the selection phenotype. Adapted from So RWL et al., Mol Neurodegener. 2019
The project is a basic research project in the field of health that will generate advanced knowledge using high-throughput, cutting-edge techniques, such as CRISPR/Cas9 screen and –omics studies. This new information has the potential to enable effective pharmacological therapy for β-haemoglobinopathies and reduce or eliminate the transfusion requirements of these patients. Such an outcome would have a tremendous impact on their quality of life, productivity and the utilization of healthcare resources of the country and especially for poorer countries with limited healthcare infrastructure.
The research will be performed primarily at the Cyprus Institute of Neurology and Genetics (CING), which is one of Cyprus leading biological research centres in terms of both facilities and personnel. The Molecular Genetics Thalassaemia Department, which will be leading the project, has been performing research on different aspects of the β-thalassaemia for the last 20 years with numerous publications and grants awarded to it. The project will be carried out in collaboration with the Bioinformatics Department of CING, the Biomedical Research Foundation of the Academy of Athens (BRFAA) and the Thalassaemia Clinics in Nicosia and Larnaca.
The project is funded by the national Research and Innovation Foundation (RIF) under the RESTART Programs 2016-2020/Excellence Hubs (EXCELLENCE/1216/0389). The current study was reviewed and successfully approved by the Cyprus National Bioethics Committee (Αρ. Φακ.: ΕΕΒΚ/ΕΠ/2018/53).
Consortium
Host Organization – The Cyprus Institute of Neurology and Genetics (CING)
- Molecular Genetics Thalassaemia Department
Prof. Marina Kleanthous, Dr. Marios Phylactides (Project Coordinator), Dr. Petros Kountouris, Dr. Anthi Demetriadou - Bioinformatics Department
Prof. George M. Spyrou, Dr. Anastasis Oulas
Partners
- The Cypriot Ministry of Health
Dr. Soteroula Christou (Thalassaemia Clinic, Arch. Makarios III Hospital)
Dr. Maria Sitarou (Thalassaemia Clinic, Larnaca General Hospital)
Foreign Research Organization
- Biomedical Research Foundation of the Academy of Athens (BRFAA)
Dr. Eleni Katsantoni

Molecular Genetics Thalassaemia Department

Dr Phylactides’ Group, Molecular Genetics Thalassaemia Department
Dr. Anthi Demetriadou, Dr. Marios Phylactides (Project Coordinator), Prof. Marina Kleanthous (Head of the Department), Dr. Savanna Andreou

0002.jpg)