Research Grants
Research Grants to the Neuroscience Department in the last 5 years

The Neuroscience Department research interests focus on investigating the cellular and molecular mechanisms causing neurological disorders such as inherited neuropathies (Charcot-Marie-Tooth Disease, CMT), multiple sclerosis, and leukodystrophies. The development of novel experimental models in our department has led to a better understanding of disease pathogenesis in demyelinating disorders like the X-linked Charcot-Marie-Tooth disease (CMT1X) and Multiple Sclerosis. We are interested in the biology of glia cells and in particular gap junctions in the peripheral and central nervous system. Novel gene therapy approaches are underway for the replacement of connexin genes that could be used to treat demyelinating neurological diseases.
The main research areas of the Neuroscience Department are:
Investigation of cellular and molecular mechanisms that lead to the manifestation of neuropathy and encephalopathy in patients with inherited mutations in gap junction proteins:
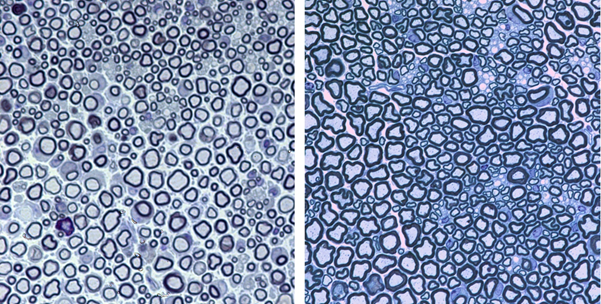
Gap junctions (GJs) are crucial for the ability of glial cells and neurons to maintain active communication and to preserve homeostasis in the functioning nervous system. Numerous mutations in gap junction proteins like the Cx32 are the major cause of CMT1X disease. Transgenic mouse models expressing Cx32 mutations in both central and peripheral myelinating cells were generated for the first time in our facilities (Sargiannidou et al.; 2009).
These transgenic mice, as well as in-vitro cell cultures, are used to investigate any trans-dominant effects of Cx32 mutations on other GJ proteins, and to examine the contribution of these mutations under conditions of metabolic and inflammatory stress in the development of CNS phenotypes in CMT1X (Sargiannidou et al.; 2008, Sargiannidou et al.; 2009, Olympiou et al.; 2016). Moreover, interference of these mutations are also observed in gene therapy (Kyriakoudi et al.; 2017, Kagiava et al.; 2018).
Furthermore, additional gap junction proteins are characterized in glia cells (Altevogt et al.; 2002, Kleopa et al.; 2004), and novel mutations are identified that could possibly be involved in disease pathogenesis (Sargiannidou et al.; 2009, Sargiannidou et al.; 2015a).
Development of gene therapy approaches for gene replacement in neuropathies and leukodystrophies:
The development of gene therapy approaches for connexin gene replacement in myelinating glial cells of the peripheral and central nervous systems is an important objective of the Neuroscience Department. Various delivery methods of lentiviral and AAV vectors harboring connexin genes are investigated in transgenic and knockout mouse models resulting in major pathological improvements. In addition, cell-targeted expression in myelinating glial cells of the peripheral and central nervous systems are explored through cell-specific and myelin-specific promoters, for a safer and more precise treatment.
Intraneural and intrathecal GJB1 (Cx32) gene delivery targeting Schwann cells in GJB1 knockout mice shows long-lasting and widespread expression of the neuropathy-associated gene (Cx32) in multiple spinal roots and peripheral nerves, as well as rescue of the peripheral neuropathy phenotype of CMT1X (Sargiannidou et al.; 2015b, Kagiava et al.; 2016, Kagiava et al, 2019). Furthermore, gene replacement therapy in a model of Charcot-Marie-Tooth 4C (CMT4C) neuropathy with SH3TC2 loss of function mutations was also successful after intrathecal SH3TC2 gene delivery (Schiza et al.; 2019). Gene therapy approaches targeting Oligodendrocytes of the CNS are investigated through different delivery methods (intracerebral, intraventricular and parenchymal injections). The Oligodendrocyte specific connexin 47 (Cx47/GJC2) is crucial for gap junctional communication throughout the central nervous system and GJC2 mutations can lead to demyelination and leukodystrophy. Targeted gene delivery, as well as transgenic expression of Cx47 or Cx32, was able to ameliorate demyelination, inflammation and astrogliosis in both the brain and spinal cord (Kagiava et al.; 2014, Schiza et al.; 2015, Georgiou et al.; 2017). More recently, AAV vector based gene therapy targeting Schwann cells and oligodendrocytes has been established to treat PNS and CNS disorders (Kagiava et al, 2021a, Kagiava et al., 2021b). Moreover, in a mouse model of CMT1A, using AAV9-miR871-driven silencing of PMP22, we managed to ameliorate the myelin pathology of the peripheral nerves (Stavrou et al., 2022).
Examination of glial connexin pathology in acquired demyelination:
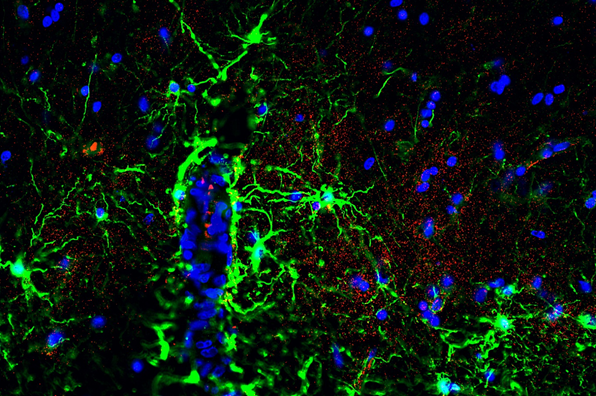
In inflammatory demyelinating disorders like Multiple sclerosis (MS), focal lesions in the central nervous system lead to demyelination, axonal loss, and astrogliosis. Research in the Neuroscience Department utilizes the experimental encephalomyelitis (EAE) mouse model of MS and connexin deficient mice, in addition to postmortem human MS brain samples to investigate the role of glial gap junctions in MS pathology.
Studies have identified widespread glial connexin pathology with loss of gap junctions in oligodendrocytes within white and gray matter lesions, but also in normal-appearing tissue (Markoullis et al.; 2012a, Markoullis et al.; 2012b). In addition, evidence of reactive astrocytosis and disconnection of astrocytes from oligodendrocytes correlate with inflammation and disease progression, emphasizing the role of connexins in MS (Markoullis et al.; 2014).
Furthermore, in connexin deficient mice with induced EAE, oligodendrocyte GJs are found to play a critical role in the regulation of inflammation and myelin loss as they could influence cytokine expression and Blood-Brain barrier integrity (Papaneophytou*, Georgiou* et al.; 2018, Stavropoulos et al., Pharmaceuticals 2021).
Most of these research projects are based on on-going collaborations with leading scientists at academic institutions in Europe and the USA, including the University of Pennsylvania, Nationwide Children’s Hospital Ohio, University of Wisconsin, University of Texas Southwestern, USA, Imperial College London, University of Oxford, and University of Cambridge, UK, and Autonomous University of Barcelona, Spain.
In the past several years, research projects in the Neuroscience department are funded by the National Multiple Sclerosis Society of the USA, the Muscular Dystrophy Association, USA (MDA), the Charcot–Marie–Tooth Association (CMTA) and CMT Research Foundation, of the USA, the European Leukodystrophy Association (ELA), the AFM-Téléthon France, the Cyprus Research & Innovation Foundation, and the Cyprus Telethon, in collaboration with scientists from renowned institutions.
Research Grants to the Neuroscience Department in the last 5 years
Selected publications of the Neuroscience Department in the last 5 years: Stavrou M, Kleopa KA. CMT1A current gene therapy approaches and promising ...
Selected Awards to Members of the Neuroscience Department in the last 5 years: • European Academy of Neurology ...

0002.jpg)